
The scientific literature is growing at an unprecedented rate, making it increasingly difficult to stay up to date with the latest research. Fortunately, various tools are available to help researchers keep pace, including PubMed notifications and Google Scholar alerts. However, these tools have limitations when it comes to discovering manuscripts that explore similar concepts. They often rely on keyword searches, which can be ineffective in capturing the nuances of a paper’s underlying ideas. When searching databases like PubMed and platforms like bioRxiv, the main challenge lies in finding manuscripts that share conceptual similarities, as these tools typically prioritize keywords over the deeper meanings of the papers.
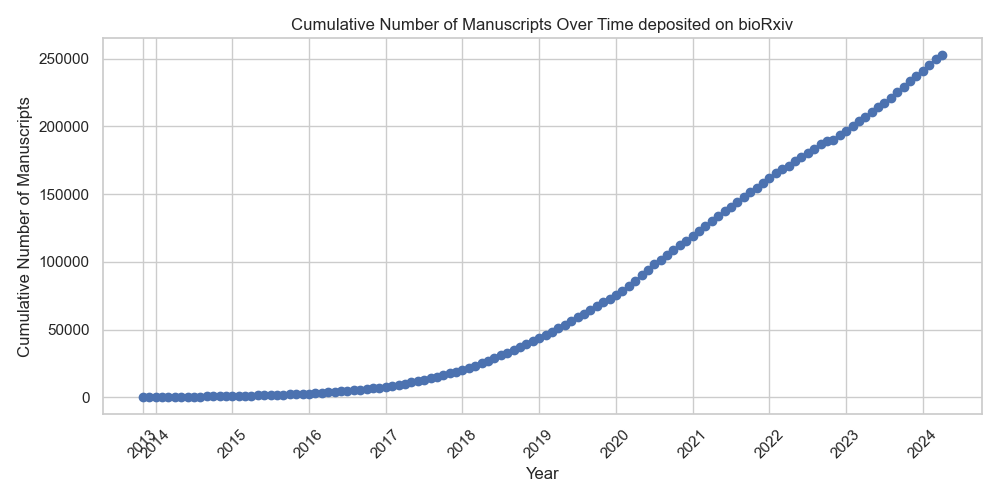
Efficiently navigating the vast and rapidly expanding space of scientific publications requires novel tools for literature search. In this post, I show how to develop a simple yet powerful tool for exploring bioRxiv manuscripts based on a search query. Semantic search represents a significant advancement for the scientific community. It enables researchers to explore texts based on their underlying meanings rather than relying solely on specific keywords. This approach facilitates a more nuanced and comprehensive literature search.
Text classification is a crucial aspect of this process, involving the categorization of text into organized groups based on its content and context. While keyword searches and topic modeling are useful methods, semantic analysis stands out for its ability to capture the deeper meaning of text.
“Recent advancements in natural language processing technologies have enabled the development of models capable of computing text embeddings. These are numerical representations that capture the core ideas and context of the text, allowing for a more sophisticated analysis of content. By leveraging models like BART and bioBERT, which excel at capturing complex semantic relationships within text, we can perform sophisticated text classification. This approach was instrumental in my exploration of research trends in cryo-electron microscopy (cryo-EM). By analyzing the semantic similarity of abstracts, I successfully grouped cryo-EM manuscripts into clusters that reflect their thematic concentrations. The results are visualized in the graph below, providing a clear representation of the predominant research themes within cryo-EM:
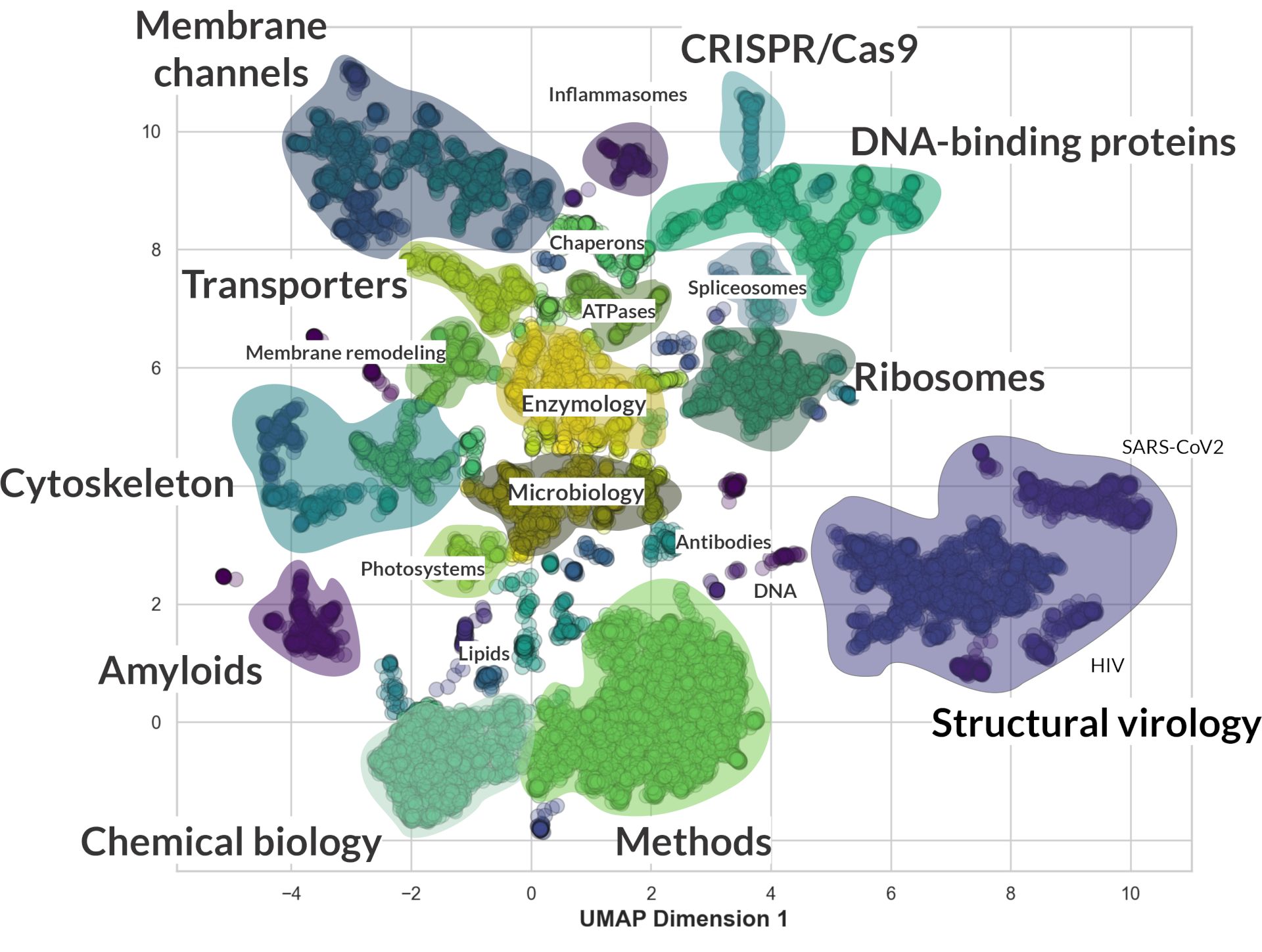
The embedding process generates multidimensional vectors, originally consisting of 2048 dimensions. These were effectively reduced to two dimensions using a technique called Uniform Manifold Approximation and Projection (UMAP), which simplifies the data while preserving its structural relationships for easier analysis and visualization. This technique allows dimension reduction by leveraging the connections and topological features among data points. This reduction enables the observation of interesting dependencies, as different topics, such as ribosomes, transcription factors, spliceosomes, and various viruses, cluster together. However, the placement of these clusters on the graph is influenced by the specific UMAP settings used; therefore, the results should be considered indicative rather than definitive, making this more of an exploratory project than a conclusive study.
Furthermore, beyond merely plotting and examining the relationships between abstracts through their embedding vectors, these vectors can be used to search for abstracts similar to a given query. The search query itself is converted into an embedding vector, and the similarity between this query vector and those of the abstracts is calculated. Abstracts are then ranked based on this similarity, and the top N abstracts are retrieved. This post, inspired by a HuggingFace blog post, will describe how to do it.
The initial step involves setting up the Python environment and fetching the data. Much of my recent programming has used tools like ChatGPT with Python focused GPT, which allows for rapid verification of code correctness, that is is contrast to possible language models halucinations in non-code content that is much more difficult to test.
bioRxiv provides a well-structured API that allows for the retrieval of basic manuscript data in JSON format. An example of a single entry is as follows:
{
"doi": "10.1101/2023.12.01.569666",
"title": "Intracellular Ebola Virus nucleocapsid assembly revealed by in situ cryo-electron tomography.",
"authors": "Watanabe, R.; Zyla, D.; Parekh, D.; Hong, C.; Jones, Y.; Schendel, S. L.; Wan, W.; Castillon, G.; Saphire, E. O.",
"author_corresponding": "Erica Ollmann Saphire",
"author_corresponding_institution": "La Jolla Institute for Immunology",
"date": "2023-12-02",
"version": "1",
"type": "new results",
"license": "cc_by_nc_nd",
"category": "microbiology",
"jatsxml": "https://www.biorxiv.org/content/early/2023/12/02/2023.12.01.569666.source.xml",
"abstract": "Filoviruses, including Ebola and Marburg viruses, are a family of highly lethal viruses that cause severe hemorrhagic fever in humans with near annual outbreaks. Antiviral options are limited, and broad-spectrum antivirals are needed. The core of the filamentous virion is formed by the nucleocapsid, a helical structure in which the polymerized nucleoprotein (NP) assembles on the RNA genome and is surrounded by viral proteins VP24 and VP351,2. The mechanism by which these proteins assemble to form nucleocapsids inside infected cells is unclear, and the identity of the outer half of nucleocapsid density remains unassigned. Using cryo-electron tomography, we revealed the assembly process of Ebola virus in both cells transfected with the viral proteins and cells infected with a biologically contained model Ebola virus. We obtained a fully assembled intracellular nucleocapsid-like structure at 9 [A] that allows assignment of previously unassigned densities and presents the first complete model of the intracellular Ebola nucleocapsid. Our model reveals a previously unknown, third layer of additional copies of NP in complex with VP35. In this outer layer, the N-terminal domain of VP35 maintains NP in a monomeric form, and the rest of VP35 appears to participate in an intra-rung nucleocapsid interaction. The C-terminal region of NP lies outside this third outer layer, allowing localization and recruitment of additional viral proteins and connection of the nucleocapsid to the matrix beneath the virus envelope. Comparison of this in-cell nucleocapsid to previous in-virion structures reveals that the nucleocapsid helix condenses upon incorporation into the virion. Molecular interfaces responsible for nucleocapsid assembly are conserved among filoviruses, offering potential target sites for broadly effective antivirals. The architecture and assembly dynamics of Ebola virus nucleocapsids revealed here uncover a unique filovirus nucleocapsid assembly and a condensation mechanism that protect viral genome integrity in the virion.",
"published": "NA",
"server": "biorxiv"
},
As evident from the data fetched, several key details, such as important keywords, are included, although the main manuscript content is not readily accessible through this method. To obtain the complete content of the manuscripts from bioRxiv, one would need to retrieve it from Amazon Web Services (AWS), which requires a fee. However, for the purposes of this demo, we will proceed with using the free API. The following code snippet allows the relatively quick retrieval of the entire bioRxiv database:
# Fast fetch data from bioRxiv
import requests
import json
import os
from datetime import datetime
from dateutil.relativedelta import relativedelta
from concurrent.futures import ThreadPoolExecutor, as_completed
def fetch_and_save_data_block(endpoint, server, block_start, block_end, save_directory, format='json'):
base_url = f"https://api.biorxiv.org/{endpoint}/{server}/"
block_interval = f"{block_start.strftime('%Y-%m-%d')}/{block_end.strftime('%Y-%m-%d')}"
block_data = []
cursor = 0
continue_fetching = True
while continue_fetching:
url = f"{base_url}{block_interval}/{cursor}/{format}"
response = requests.get(url)
if response.status_code != 200:
print(f"Failed to fetch data for block {block_interval} at cursor {cursor}. HTTP Status: {response.status_code}")
break
data = response.json()
fetched_papers = len(data['collection'])
if fetched_papers > 0:
block_data.extend(data['collection'])
cursor += fetched_papers # Update the cursor to fetch next set of data
print(f"Fetched {fetched_papers} papers for block {block_interval}. Total fetched: {cursor}.")
else:
continue_fetching = False
if block_data:
save_data_block(block_data, block_start, block_end, endpoint, save_directory)
def save_data_block(block_data, start_date, end_date, endpoint, save_directory):
start_yymmdd = start_date.strftime("%y%m%d")
end_yymmdd = end_date.strftime("%y%m%d")
filename = f"{save_directory}/{endpoint}_data_{start_yymmdd}_{end_yymmdd}.json"
with open(filename, 'w') as file:
json.dump(block_data, file, indent=4)
print(f"Saved data block to {filename}")
def fetch_data(endpoint, server, interval, save_directory, format='json'):
os.makedirs(save_directory, exist_ok=True)
start_date, end_date = [datetime.strptime(date, "%Y-%m-%d") for date in interval.split('/')]
current_date = start_date
tasks = []
with ThreadPoolExecutor(max_workers=12) as executor: # Adjust the number of workers as needed
while current_date <= end_date:
block_start = current_date
block_end = min(current_date + relativedelta(months=1) - relativedelta(days=1), end_date)
tasks.append(executor.submit(fetch_and_save_data_block, endpoint, server, block_start, block_end, save_directory, format))
current_date += relativedelta(months=1)
for future in as_completed(tasks):
future.result()
endpoint = "details"
server = "biorxiv"
interval = "2010-03-24/2025-03-31"
save_directory = "biorxiv_data" # Specify your custom directory here
fetch_data(endpoint, server, interval, save_directory)
This code allows fetching the whole bioRxiv abstract database and save it into JSON files. While JSON is a versatile format, for simplicity and perhaps due to familiarity, I prefer using pandas for handling database-related tasks. Therefore, the next section of the code is designed to convert the fetched JSON files into a pandas DataFrame format. This transformation is followed by saving the data as Parquet files, which are significantly faster to read and write compared to JSON:
import pandas as pd
import os
import json
from pathlib import Path
def load_json_to_dataframe(json_files):
"""Load and aggregate JSON data from a list of files into a pandas DataFrame."""
all_data = []
for file_path in json_files:
with open(file_path, 'r') as file:
data = json.load(file)
all_data.extend(data)
return pd.DataFrame(all_data)
def save_dataframe(df, save_path, file_num):
"""Save DataFrame to a file in Parquet format."""
df.to_parquet(f"{save_path}_part_{file_num}.parquet")
def process_json_files(directory, save_directory, n=10):
"""Process all JSON files in a directory and save aggregated data every N files."""
# Ensure the save directory exists
os.makedirs(save_directory, exist_ok=True)
json_files = list(Path(directory).glob('*.json'))
total_files = len(json_files)
file_num = 0
for i in range(0, total_files, n):
# Process in batches of N files
batch_files = json_files[i:i+n]
df = load_json_to_dataframe(batch_files)
# Save the aggregated data from N files
save_dataframe(df, os.path.join(save_directory, 'aggregated_data'), file_num)
file_num += 1
print(f"Processed and saved batch {file_num} (files {i+1}-{min(i+n, total_files)})")
directory = r'biorxiv_data' # Directory containing JSON files
save_directory = r'reduced_dataframe' # Directory to save aggregated data
process_json_files(directory, save_directory, n=5)
Having the database ready we can easily check the statistics related to the fecthed manuscripts, such as category distribution:
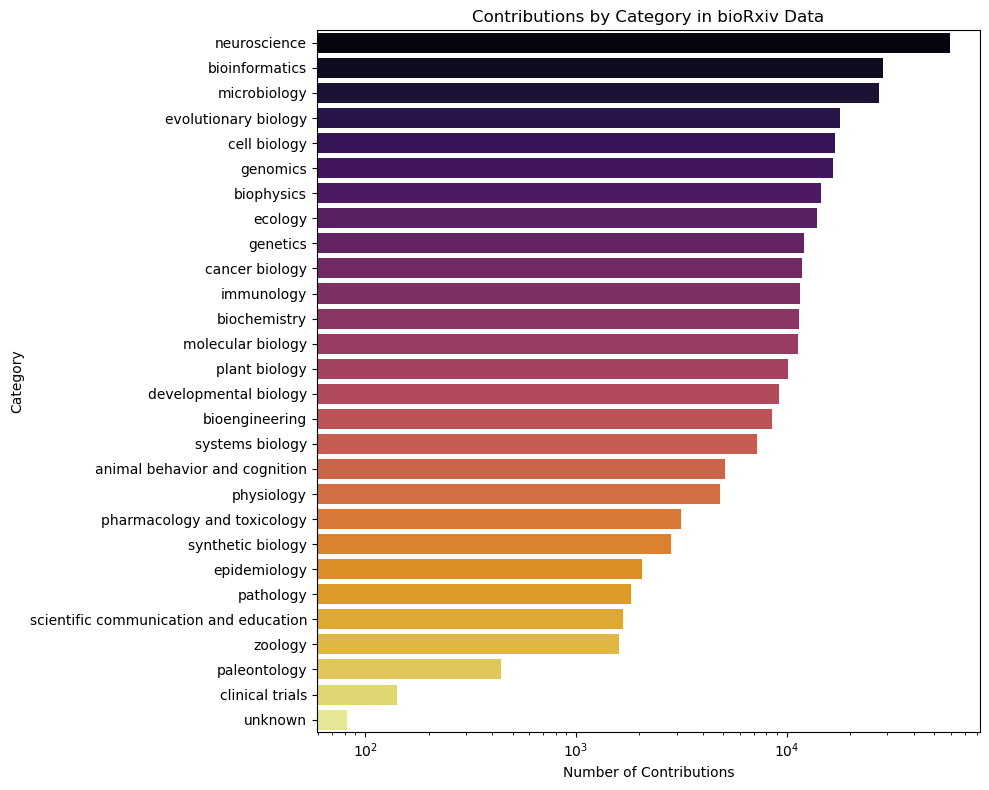
Surprisingly, most of the manuscripts deposited on bioRxiv are related to neurobiology. There are few with unknown tags, which might be a good idea to check.
With the database now prepared, we can move forward to calculate the embeddings. In this endeavor, I have decided to adhere closely to the methodology outlined in a HuggingFace blog post, albeit with some minor modifications. Typically, embeddings are calculated using the float32 data type, which allows for a wide range of distinct values (2^32), encompassing a substantial amount of information. An effective strategy to manage data more efficiently without losing significant information is to use quantized vectors. By converting float32 to float16, the amount of data required is halved while maintaining a comparable level of information. Further reductions can be achieved by using int8 or binary precision, which are particularly advantageous in production environments where speed and memory usage are critical constraints. It’s worth noting that reducing the embeddings from float32 to int8 retains approximately 96% of the precision, an acceptable compromise considering the significantly reduced space required by the embeddings.
For the computation of embeddings, I used the mixedbread-ai/mxbai-embed-large-v1 model from HuggingFace, used with normalization and set to ubinary precision:
import pandas as pd
import numpy as np
from pathlib import Path
import datetime
import requests
import json
import os
from datetime import datetime
from dateutil.relativedelta import relativedelta
from concurrent.futures import ThreadPoolExecutor, as_completed
import pandas as pd
import os
import json
from pathlib import Path
from sentence_transformers import SentenceTransformer, models
import torch
import shutil
def load_data_embeddings(existing_data_path):
# Load existing database and embeddings
df_existing = pd.read_parquet(existing_data_path)
# create a mask for filtering
mask = ~df_existing.duplicated(subset=["title"], keep="last")
df_existing = df_existing[mask]
return df_combined
unprocessed_dataframes = load_data_embeddings()
if unprocessed_dataframes:
for file in unprocessed_dataframes:
df = pd.read_parquet(file)
query = df['abstract'].tolist()
device = "cuda" if torch.cuda.is_available() else "cpu"
model = SentenceTransformer("mixedbread-ai/mxbai-embed-large-v1")
model.to(device)
# this will likely crash due to memory limits
query_embedding = model.encode(query, normalize_embeddings=True, precision='ubinary', show_progress_bar=True)
file_path=os.path.basename(file).split('.')[0]
embeddings_path = f'embed_update/{file_path}'
np.save(embeddings_path, query_embedding)
print(f'Saved embeddings {embeddings_path}')
# remove old json directory
db_update_json = 'db_update_json'
shutil.rmtree(db_update_json)
print(f"Directory '{db_update_json}' and its contents have been removed.")
else:
print('Nothing to do')
When processing over 250,000 abstracts on my NVIDIA GeForce RTX 2080 Ti, it took approximately three hours. While this task can be accomplished on a single machine, larger databases may require the use of a distributed system to achieve efficient processing times. Once the embeddings are calculated and saved, we can proceed with building the search tool using Facebook’s FAISS library. This powerful tool is optimized for high-dimensional vectors and is widely used in production environments due to its efficiency and scalability.
The following code snippet demonstrates how to perform semantic search using FAISS:
import faiss
from sentence_transformers.quantization import quantize_embeddings, semantic_search_faiss
# Specify the target precision for the corpus embeddings
corpus_precision = "ubinary"
queries = [
'new cryo-EM freezing technique',
'cryo-EM spray'
]
corpus_index = None
# Encode the queries
start_time = time.time()
query_embeddings = model.encode(queries, normalize_embeddings=True, precision='ubinary')
print(query_embeddings)
print(f"Encoding time: {time.time() - start_time:.6f} seconds")
# Perform semantic search using FAISS
results, search_time, corpus_index = semantic_search_faiss(
query_embeddings,
corpus_index=corpus_index,
corpus_embeddings=embeddings_unique if corpus_index is None else None,
corpus_precision=corpus_precision,
top_k=10,
calibration_embeddings=None, # Adjust based on your setup for uint8
rescore=corpus_precision != "float32",
rescore_multiplier=4,
exact=True,
output_index=True,
)
# Output the results
print("Precision:", corpus_precision)
print(f"Search time: {search_time:.6f} seconds")
for query, result in zip(queries, results):
print(f"Query: {query}")
for entry in result:
print(f"(Score: {entry['score']:.4f}) {df_unique['title'].tolist()[entry['corpus_id']]}")
print("")
Given code produces this output:
query_embeddings=
[[213 72 128 76 142 242 59 222 213 138 113 223 119 146 92 187 7 107
77 116 196 247 255 153 64 83 100 123 83 127 181 92 197 232 43 25
217 5 125 94 36 132 123 100 242 242 181 239 83 14 251 151 17 174
16 7 136 166 78 233 113 41 161 0 49 18 96 192 19 101 92 174
17 176 48 104 248 102 52 84 130 252 56 234 32 5 168 175 157 190
245 75 172 1 57 185 206 39 167 191 216 113 140 234 226 101 159 116
190 119 102 3 221 152 136 143 124 28 178 178 109 146 212 22 184 123
71 51]
[209 9 144 77 174 241 120 94 213 33 125 215 127 178 77 163 14 105
253 60 117 39 251 131 86 67 114 3 149 126 197 92 149 212 11 141
81 6 205 94 68 133 107 104 243 227 21 99 81 131 251 135 80 175
88 39 137 226 79 45 97 105 161 53 49 26 97 193 23 101 94 174
26 177 136 44 216 102 116 244 131 253 48 160 2 6 25 109 141 190
213 218 174 5 49 184 136 63 175 28 162 49 157 106 234 85 151 120
230 55 164 19 207 152 140 135 117 31 162 213 77 178 212 214 152 222
194 48]]
Encoding time: 0.026252 seconds
Precision: ubinary
Search time: 0.004001 seconds
Query: new cryo-EM freezing technique
(Score: 176.0000) An open-source cryo-storage solution
(Score: 178.0000) Routine Determination of Ice Thickness for Cryo-EM Grids
(Score: 182.0000) Factors affecting macromolecule orientations in thin films formed in cryo-EM
(Score: 182.0000) Direct information estimation from cryo-EM Movies with CARYON
(Score: 184.0000) Vitrification after multiple rounds of sample application and blotting improves particle density on cryo-electron microscopy grids.
(Score: 184.0000) Shake-it-off: A simple ultrasonic cryo-EM specimen preparation device
(Score: 185.0000) HPM Live μ for full CLEM workflow
(Score: 186.0000) A streamlined workflow for automated cryo focused ion beam milling
(Score: 187.0000) AgarFix: simple and accessible stabilization of challenging single-particle cryo-EM specimens through cross-linking in a matrix of agar
(Score: 187.0000) Automated cryo-EM sample preparation by pin-printing and jet vitrification
Query: cryo-EM spray
(Score: 226.0000) Shake-it-off: A simple ultrasonic cryo-EM specimen preparation device
(Score: 232.0000) Through-grid wicking enables high-speed cryoEM specimen preparation
(Score: 233.0000) Automated cryo-EM sample preparation by pin-printing and jet vitrification
(Score: 236.0000) A case for glycerol as an acceptable additive to cryoEM samples
(Score: 237.0000) Theoretical framework and experimental solution for the air-water interface adsorption problem in cryoEM
(Score: 239.0000) Application of monolayer graphene to cryo-electron microscopy grids for high-resolution structure determination
(Score: 239.0000) CryoCycle your grids: Plunge vitrifying and reusing clipped grids to advance cryoEM democratization
(Score: 239.0000) Effect of charge on protein preferred orientation at the air-water interface in cryo-electron microscopy
(Score: 239.0000) A mixture of innate cryoprotectants is key for cryopreservation of a drosophilid fly larva.
(Score: 239.0000) A mixture of innate cryoprotectants is key for freeze tolerance and cryopreservation of a drosophilid fly larva.
As you can see, the search results accurately match the query terms, and the search process itself is remarkably efficient. In fact, searching over 250,000 abstracts takes only 0.004 seconds, an impressive performance that demonstrates the power of our semantic search tool with FAISS. Moreover, even with a database 200 times larger, we would still be able to retrieve relevant results in under 1 second, assuming linear time complexity. Furthermore, the quality of the retrieved abstracts is also excellent, as they are closely related to the original query.
To further enhance its capabilities, the search tool can be improved by adding advanced features such as filtering by publication date, author, or category. Additionally, the tool can be integrated into a web application to provide a user-friendly interface for exploring bioRxiv manuscripts. For this purpose, I’ve combined all previous code into a Streamlit app that’s accessible on GitHub or 🤗 Hugging Face.
Recently, the hype surrounding large language models has been peaking. I’ve decided to add support for abstract summaries using Groq’s Language Processing Unit (LPU), which enables extremely fast inference – over 500 tokens per second, nearly 10-fold faster than OpenAI. This summary of the first 10 abstracts provides a valuable glimpse into what lies within. The prompt used in this case looks like this:
Review the abstracts listed below and create a list and summary that captures their main themes and findings. Identify any commonalities across the abstracts and highlight these in your summary. Ensure your response is concise, avoids external links, and is formatted in markdown.\n\n
The tool is quite powerful and can be used for various purposes, from simple search to advanced analysis. It’s also super fast on shared resources like Streamlit Cloud or Hugging Face Spaces, and can even work faster using local machine. The functionality could be expanded by obtaining full publication embeddings divided into abstracts, results, discussion, and methods. However, due to limited resources and money, I won’t be able to do that, but I hope the code will be useful for someone who wants to expand it.
What’s next? There are several databases that could greatly benefit from semantic search, including PubMed and PDB. The current search functionality on PDB leaves much to be desired, with searches for glycoprotein returning random entries and sorting searches affecting all entries, rather than just the query results. A semantic search tool could revolutionize the way structural biologists interact with this database by providing a more intuitive and efficient way of searching and exploring the data.
One potential approach would be to use embeddings for PDB entry information, related UniProt data, and relevant publications abstracts or full text. By combining these elements, we could create a powerful tool that would greatly enhance the search experience. However, as the data needs to be fetched independently, this poses a significant undertaking.
On the other hand, PubMed presents a different challenge due to its massive library of ~40M abstracts, which is a significant hurdle for my current hardware. The PubMed abstracts related XML files have ~370 GB, curated Parquet files with abstract and basic information are ~19 GB, and quantized embeddings would require 4.7 GB of RAM space. While this is not beyond the capabilities of a normal computer, it would impose significant limitations on an online app.
We can create a simple tool that helps scientists quickly find what they need in the vast amount of scientific literature. Having all relevant papers in one place, along with brief summaries, could lead to new ideas and insights. Who knows exactly what might come from it, but it’s definitely worth trying.